Mechanisms that regulate tau spread
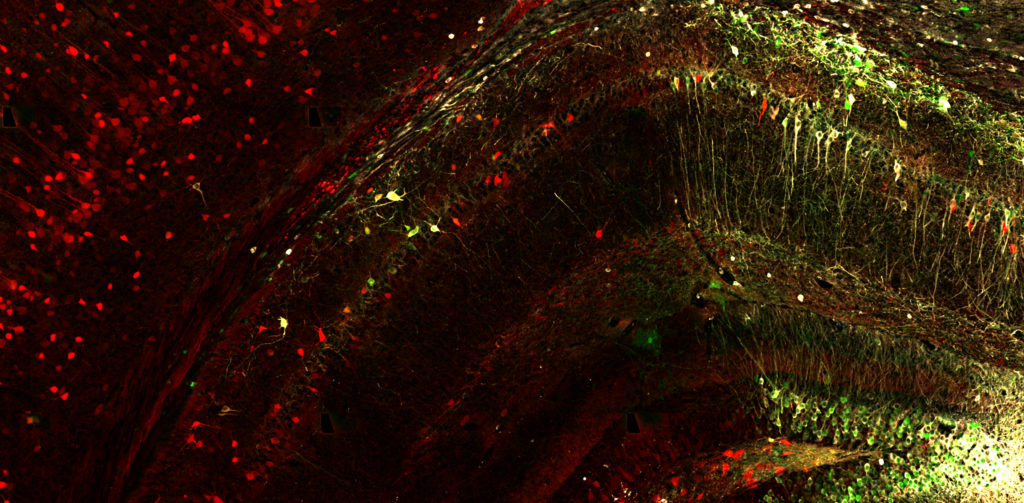
The spread and aggregation of the microtubule-associated protein tau has been linked to a variety of neurodegenerative diseases, collectively known as tauopathies. Mounting evidence suggests that the spread of tau aggregates may be causative for disease, however the cellular mechanism(s) that facilitate tau spread are still unclear. Recently, we identified a cellular receptor, LRP1, that can regulate tau entry into the cell. This discovery has spurred many exciting questions; what are the molecular features that define the tau-LRP1 interaction? How is this interaction exploited during disease? Can this protein-protein interaction be targeted for disease therapeutics?
The role of cellular identity in disease
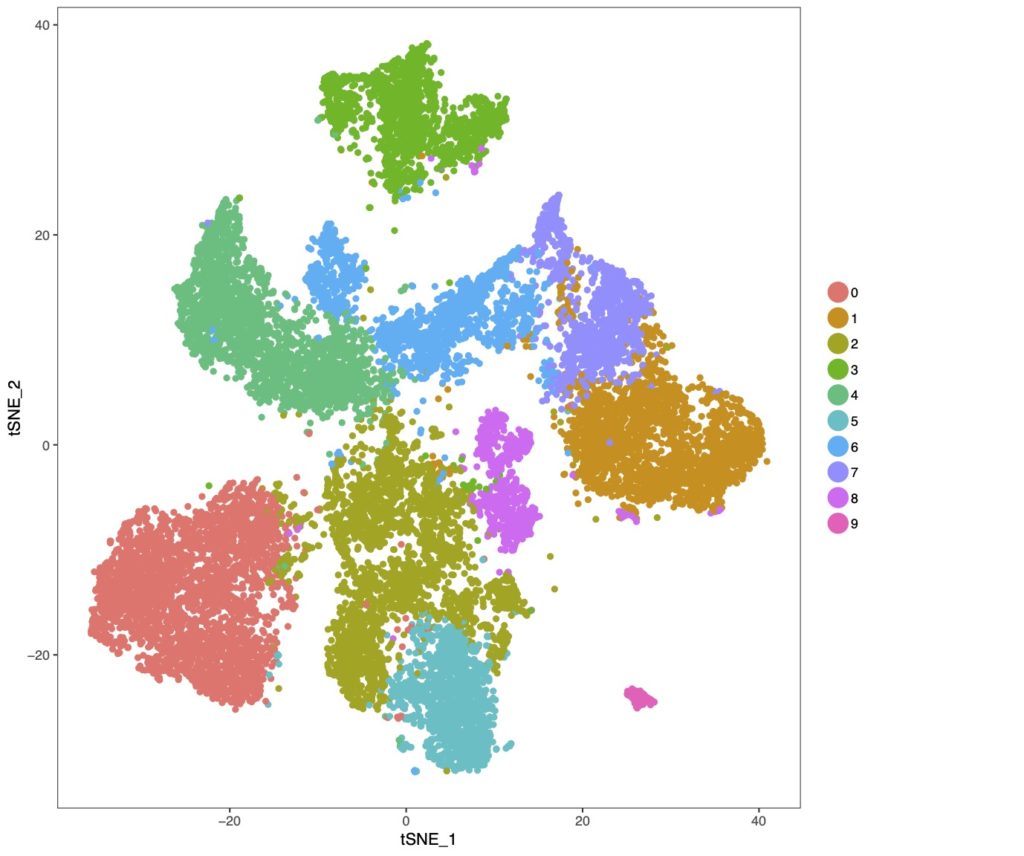
There is an increasing amount of appreciation that tauopathy progression is not solely a consequence of neuronal dysfunction, but may also be mediated by glia-dependent neuroinflammatory mechanisms. Microglia and astrocytes are the major glial subtypes that respond to disease stressors using the innate immune response (production and release of inflammatory mediators). Microglia have been historically viewed as mainly protecting the brain from exogenous insults, and astrocytes were seen as primarily providing nutritive and structural support. However, both cell types are now known to play important roles in brain health, and in neurodegenerative disease their function may be adversely affected through inflammatory signaling responses. To understand this complex phenotype, we use state-of-the-art single cell transcriptomic technologies to uncover how cellular identity influences cellular response mechanisms. This strategy allows us to understand how cell types interface with one another to function in disease.
Understanding tau’s physical state
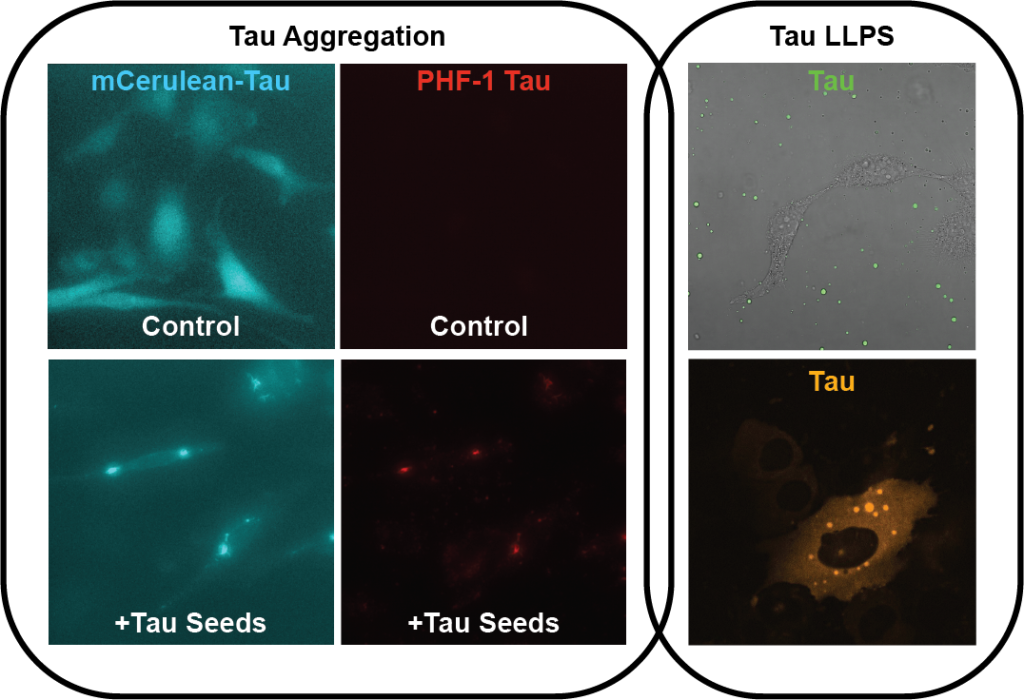
Tau is an abundant and highly soluble protein, yet is known to aggregate in tauopathies. Tau is an intrinsically disordered protein (IDP), and various methods have determined it to be largely random coil in solution. During aggregation, however, tau adopts a cross β-sheet structure (reminiscent of other amyloids) that allows non-native interactions between monomers leading to the formation of amorphous oligomers and eventually long fibrils. While the aggregation of tau is a known correlative with disease, the mechanisms leading to filament formation inside the cell have yet to be identified. In contrast to its aggregation, it has recently been discovered that tau can also undergo a very different structural transition, namely from a soluble diffuse form to a liquid-liquid phase separated (LLPS) form. The LLPS of tau is similar to other IDPs (FUS, TDP43, hnRNPA1, etc.) and recent works have started to speculate that this phase separated version of tau may be on pathway to its aggregation state. In the lab, we interrogate the cellular mechanism(s) that control tau’s conversion to insoluble aggregates within cells, and try to understand if these pathways converge with tau phase separation mechanisms.